
PIERWOTNE CHŁONIAKI SKÓRY
- ang. Primary cutaneous lymphomas
- międzynarodowy skrót: CLs
Mianem pierwotnych chłoniaków skóry określa się nowotwory układu limfatycznego, które w momencie diagnozy ograniczone są wyłącznie do skóry i nie stwierdza się zmian w węzłach chłonnych, szpiku czy narządach wewnętrznych. Około 65% chłoniaków skóry wywodzi się z komórek T (CTLC, cutaneous T-cell lymphoma), 25% z komórek B (CBCL, cutaneous B-cell lymphoma), a pozostałe 10% z komórek NK (NK, natural killers).
Obecnie stosowaną klasyfikacją pierwotnych chłoniaków skóry jest zaktualizowana klasyfikacja WHO-EORTC z 2018 r.
DIAGOSTYKA
Postępowanie diagnostyczne w przypadku podejrzenia chłoniaka skóry obejmuje:
- Badanie podmiotowe i przedmiotowe oceniające morfologię zmian skórnych, badanie węzłów chłonnych, wątroby i śledziony
- Badania obrazowe, które w zależności od rodzaju i stadium nowotworu mogą obejmować ultrasonografię, RTG klatki piersiowej, TK klatki piersiowej, brzucha, miednicy, MR brzucha i miednicy
- Badania laboratoryjne, takie jak morfologia krwi obwodowej z rozmazem, immunofenotypowanie krwi obwodowej, immunoelektroforeza osocza, ocena funkcji nerek i wątroby, stężenie IgE całkowitego, dehydrogenazy mleczanowej i kwasu moczowego, a w niektórych przypadkach także badanie szpiku kostnego (trepanobiopsja oraz biopsja aspiracyjna)
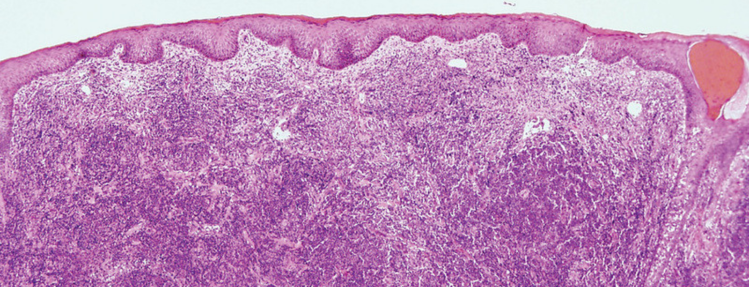
- Badanie histopatologiczne – w większości przypadków diagnoza pierwotnego chłoniaka skóry stawiana jest na podstawie badania histopatologicznego wycinka skóry pobranego w trakcie biopsji skóry. Procedura ta jest wykonywana w większości ośrodków dermatologicznych. Do pobrania biopsji wykorzystuje się 4-mm sztancę, a zabieg przeprowadzany jest w znieczuleniu miejscowym.
Przez 2-4 tygodnie przed planowaną biopsją pacjenci nie powinni stosować leków miejscowych, takich jak np. maści glikokortykosteroidowe. Istotnym elementem badania histopatologicznego jest ocena immunofenotypową nieprawidłowych komórek (immunohistochemia i immunocytochemia), która wraz z badaniami cytogenetycznymi i molekularnymi umożliwia właściwą klasyfikację nowotworu zgodnie z klasyfikacją WHO-EORTC.W części przypadków analiza dokonywana jest na podstawie oceny węzła chłonnego lub fragmentu innego zajętego narządu.
CHŁONIAKI Z DOJRZAŁYCH KOMÓREK T I NK
Ziarniniak grzybiasty
Ziarniniak grzybiasty (MF, mycosis fungoides) jest najczęstszym chłoniakiem skóry wywodzącym się z dojrzałych komórek T i cechuje się przewlekłym, stopniowo postępującym przebiegiem. Częstość występowania choroby ocenia się na 0,3-0,5:100 000 osób. Zachorowania dotyczą głównie ludzi dorosłych, częściej mężczyzn, ze szczytem zachorowań w wieku 55-60 lat.
OBJAWY
Obraz kliniczny charakteryzuje się dużą różnorodnością objawów, a obecne zmiany skórne mogą być niecharakterystyczne. Ich spektrum obejmuje zarówno zlokalizowane lub rozsiane plamy i blaszki, jak również uogólnione zajęcie skóry (erytrodermię). Poza obecnością zmian skórnych najczęściej zgłaszanym przez pacjentów objawem jest świąd. Klasycznie wyróżnia się trzy okresy zmian:
- Okres wstępny – obejmuje okrągłe, owalne lub różnokształtne rumieniowe plamy lub blaszki barwy ciemnofioletowej z obecnym złuszczaniem naskórka o różnym nasileniu, najczęściej zlokalizowane w obrębie tułowia i pośladków. Zmianom towarzyszy świąd o różnym nasileniu. Do rzadko występujących zmian należą zmiany brodawkowate, pęcherzykowe, krostkowe lub imitujące zapalenie naczyń. Okres wstępny stanowi najdłuższą fazę choroby – może trwać wiele, a u części pacjentów progresja może nie wystąpić.
- Okres naciekowy – zmiany stają się większe, grubsze, uniesione i dobrze odgraniczone od otoczenia. Przybierają barwę od czerwonosinawej do czerwonobrązowej. Dodatkowymi objawami mogą być łysienie, powiększenie węzłów chłonnych oraz nasilony świąd. U części pacjentów dochodzi do uogólnienia zmian skórnych i rozwoju erytrodermii.
- Okres guzowaty – charakteryzuje się występowaniem szybko rosnących guzów obecnych także na skórze wcześniej niezmienionej. Cechą charakterystyczną w tej fazie choroby jest tzw. twarz lwia (facies leonina). Ponadto możliwa jest progresja do choroby układowej (powiększenie węzłów chłonnych, zajęcie szpiku kostnego i narządów – węzłów chłonnych, płuc, śledziony, wątroby, przewodu pokarmowego).
Rzadkie postaci MF to m.in. MF d’emblee charakteryzujący się gwałtownym wystąpieniem dużych guzów bez obecności wcześniejszych stadiów MF, postać erytrodermiczna, mieszkowa, ziarniniakowa, siatkowica pagetoidalna czy ziarniniakowe zwiotczenie skóry.
DIAGNOSTYKA
Podstawą rozpoznania ziarniniaka grzybiastego jest badanie histopatologiczne wycinka skóry. Obraz histopatologiczny zależy od stadium choroby – w fazie wstępnej nie stwierdza się typowych cech MF, co utrudnia postawienie właściwej diagnozy. Z tego powodu, u pacjentów prezentujących zmiany skórne mogące odpowiadać wykwitom MF, może być wskazane pobranie jednorazowo kilku wycinków lub regularne powtarzanie biopsji. W późniejszym etapie obraz histopatologiczny staje się bardziej charakterystyczny i charakteryzuje się obecnością monoklonalnej proliferacji limfocytów T pomocniczych. W przypadku podejrzenia MF niezbędnym elementem badania histopatologicznego jest immunofenotypowanie, które pozwala ustalić podtyp nowotworu.
U każdego pacjenta należy wykonać morfologię krwi z rozmazem, a także podstawowe badania biochemiczne.
W przypadkach z nasilonymi zmianami skórnymi, zajęciem węzłów chłonnych i/lub narządów wewnętrznych należy przeprowadzić pełną diagnostykę obrazową celem oceny stopnia zaawansowania.
LECZENIE
Głównym celem leczenia jest łagodzenie objawów towarzyszących MF, do których należą świąd, ból, czy pieczenie. Poza powyższym leczenie ma na celu hamowanie progresji choroby i redukcja rozległości zmian skórnych.
W okresie wstępnym zastosowanie znajdują wysokiej mocy glikokortykosteroidy miejscowe (np. propionian klobetazolu), fototerapia UVB 311 nm, a przy braku skuteczności fototerapia PUVA.
W stadium IA (rumień i naciek zajmujący <10% powierzchni ciała), IB (rumień i naciek zajmujący >10% powierzchni ciała) oraz IIA (skóra jak w stadium I z limfadenopatią bez cech histopatologicznych chłoniaka), a więc w okresie naciekowym choroby, stosuje się glikokortykosteroidy miejscowe, fototerapię UVB 311 nm (w przypadku zmian rumieniowych), PUVA-terapię (w przypadku zmian rumieniowo-naciekowych), miejscowo radioterapię na pojedyncze ogniska MF oraz mechloretaminę – roztwór 10–20 mg/ml oraz żel lub maść 0,02% podawane raz dziennie. Drugą linią leczenia jest miejscowo stosowany żel beksaroten w stężeniu 1% oraz leczenie ogólne: IFN-α, metotreksat w małych dawkach (5–25 mg tygodniowo), retinoidy (izotretynoina/acytretyna/beksaroten). W przypadku przeciwwskazań do PUVA-terapii, należy rozważyć terapię TSEB (TSEB, total skin electron beam radiation – radioterapia szybkimi elektronami na całą powierzchnię skóry). W niektórych przypadkach, z uwagi na małe ryzyko progresji (10% w ciągu 10 lat) i czas przeżycia podobny jak w populacji ogólnej, dopuszcza się postawę wyczekującą bez leczenia pod warunkiem ścisłej, regularnej obserwacji.
W stadium guzowatym (stadium IIB) stosuje się terapię systemową: retinoidy lub IFN-α, także w skojarzeniu z PUVA-terapią, TSEB, metotreksatem.
U pacjentów z erytrodermią (stadium IIIA-IIIB) można rozważyć fotoforezę pozaustrojową (extracorporeal photopheresis – ECP) w połączeniu z retinoidami, IFN-α, PUVA lub innymi terapiami ogólnymi.
Ostatnim wyborem u pacjentów w stadiach (IIB-IIIB) jest monochemioterapia (gemcitabina, pegylowana liposomalna doksorubicyna) lub polichemioterapia (CHOP), radioterapia dodana do leczenia ogólnego w skojarzeniu z leczeniem ukierunkowanym na skórę lub allogeniczny przeszczep komórek krwiotwórczych.
W przypadku zajęcia węzłów chłonnych i/lub narządów wewnętrznych (stadium IV) kryteria odpowiedzi i monitorowanie przebiegu choroby są typowe dla innych nowotworów z dojrzałych limfocytów. W leczeniu stosuje się chemioterapię (gemcitabina, pegylowana liposomalna doksorubicyna, CHOP i inne, podobne do CHOP, złożone chemioterapie), radioterapię (TSEB izolowana lub w skojarzeniu ze złożonym leczeniem ogólnym), alemtuzumab lub allogeniczny przeszczep komórek krwiotwórczych.
Chłoniak z komórek T tkanki podskórnej typu zapalenia tkanki podskórnej
Chłoniak z komórek T tanki podskórnej typu zapalenia tkanki podskórnej (subcutaneous panniculitis-like T-cell lymphoma, SPTLC) jest rzadkim nowotworem stanowiącym <1% chłoniaków skóry. Występuje równie często u kobiet i mężczyzn, najczęściej u osób w średnim wieku. Cechuje się obecnością zmian o charakterze zapalenia tkanki podskórnej ze spoistymi rumieniowymi naciekami w tkance podskórnej, zlokalizowanych głównie na kończynach dolnych i tułowiu.
Zmianom mogą towarzyszyć objawy ogólne, takie jak gorączka, zmęczenie, spadek masy ciała, cytopenie, wzrost stężenia transaminaz. U niektórych pacjentów dochodzi do rozwoju zespołu hemofagocytarnego, co jest złym czynnikiem rokowniczym. W badaniu histopatologicznym naciek chłoniaka obejmuje tkankę podskórną (nie zajmuje skóry właściwej) i jest złożony z limfocytów cytotoksycznych o fenotypie αβ CD3+CD4–CD8+ oraz licznych makrofagów. W leczeniu stosuje się glikokortykosteroidy ogólne, a w przypadkach opornych także leki immunosupresyjne (metotreksat) i polichemioterapię.
Lymphomatoid papulosis
Lymphomatoid papulosis jest chłoniakiem cechującym się obecnością nacieków złożonych z anaplastycznych limfocytów T CD30+. Występuje zarówno u dzieci, jak i u osób starszych (mediana wieku to 45 lat), częściej u mężczyzn. Zmianami skórnymi są liczne guzki i grudki na skórze tułowia i kończyn, na powierzchni których obecne są łuski, zmiany krwotoczne lub strupy. Cechą charakterystyczną jest powstawanie zmian i ich samoistne ustępowanie co ok. 3-12 tygodni.
Choroba jest przewlekła, a rokowanie w większości przypadków dobre (10-letnie przeżycie to prawie 100%). W leczeniu stosuje się kąpiele PUVA, fototerapię UVB 311 nm, retinoidy systemowe oraz niskie dawki metotreksatu (5-20 mg/tydzień). Uporczywe zmiany można usunąć chirurgicznie lub za pomocą radioterapii. Z uwagi na możliwą transformację w inne choroby limfoproliferacyjne (np. ALCL, MF, chłoniak Hodgkina), chorzy powinni pozostawać pod stałą obserwacją.
Pierwotny skórny chłoniak anaplastyczny z dużych komórek
Pierwotny skórny chłoniak anaplastyczny z dużych komórek (primary cutaneous anaplastic large cell lymphoma, ALCL) dotyczy zarówno dzieci jak i dorosłych. Głównym objawem jest obecność jednego lub kilku szybko rosnących guzów mających tendencję do wrzodzenia. Ok. 1/3 przypadków ulega samoistnemu zanikowi. U nielicznych pacjentów dochodzi do zajęcia węzłów chłonnych – w tej sytuacji należy wykluczyć układową postać ALCL. W badaniu histopatologicznym skóry stwierdza się nacieki limfocytów T pomocniczych CD30+ z różnorodną ekspresją CD3 i CD4.
Leczenie polega na chirurgicznej resekcji lub radioterapii w przypadku pojedynczych guzów, a w przypadku postaci układowej stosuje się immunoterapię, IFN-α, beksaroten, metotreksat oraz chemioterapię CHOP.
Pierwotny skórny chłoniak z małych/średnich komórek T CD4+
Pierwotny skórny chłoniak z małych/średnich komórek T CD4+ charakteryzuje się obecnością małego, wolno rosnącego guzka barwy czerwonobrązowej lub o zabarwieniu skóry, bez owrzodzenia, występującego zwykle w górnej części ciała.
W badaniu histopatologicznym wycinka skóry występuje monomorficzny naciek z małych lub średnich limfocytów o fenotypie CD3+, CD4+, CD8-, CD30+. Ponadto w nacieku mogą być obecne eozynofile oraz odczyn ziarniniakowy. Jednostka ta charakteryzuje się korzystnym rokowaniem, szczególnie w przypadku pojedynczych guzów – wówczas w leczeniu stosuje się radioterapię i leczenie chirurgiczne. W przypadku zmian rozsianych stosuje się cyklofosfamid, IFN-α lub chemioterapię.
Zespół Sézary’ego
Zespół Sézary’ego jest rzadkim zespołem, który występuje zwykle u mężczyzn w 6. lub 7, dekadzie życia. Objawy początkowo są niecharakterystyczne i mogą być mylone z innymi chorobami skóry – na skórze występują plamy rumieniowo-złuszczające, nieobejmujące całej powierzchni skóry. Następnie proces chorobowy zajmuje całą skórę, wskutek czego dochodzi do erytrodermii. Skóra jest pogrubiona, obecna jest hiperpigmentacja, złuszczanie naskórka, pogrubienie rysów twarzy (twarz lwia, facies leonina), hiperkeratoza naskórka dłoni i podeszew, dystrofia paznokci.
Zmianom towarzyszą objawy ogólne – dreszcze, uczucie zimna. Cechą charakterystyczną zespołu jest uogólnione powiększenie węzłów chłonnych. W badaniu histopatologicznym stwierdza się obecność atypowych limfocytów o mózgopodobnych jądrach, znanych jako komórki Sézary’ego lub Lutznera. Ich obecność stwierdza się także w węzłach chłonnych i krwi obwodowej. Rokowanie jest zwykle niekorzystne – średni okres przeżycia wynosi zwykle 2-4 lata. W leczeniu stosuje się fotoforezę pozaustrojową, często w połączeniu z beksarotenem lub IFN-α, metotreksat, chemioterapię, gemcytabiną lub liposomalną doksorubicyną, czy allo-HSCT.
Pierwotny skórny chłoniak z komórek T γδ
Pierwotny skórny chłoniak z komórek T γδ jest bardzo rzadkim nowotworem, charakteryzującym się obecnością rozsianych blaszek lub guzków występujących głównie na kończynach dolnych. Zmianom mogą towarzyszyć zajęcie błon śluzowych oraz zespół hemofagocytarny.
W badaniu histopatologicznym stwierdza się nacieki z komórek o fenotypie cytotoksycznym: CD2+, CD3+, CD5–, CD7+/–, CD56+, bF1–, γ/δ+ oraz CD4– i CD8–. Przebieg choroby jest zwykle szybko postępujący. W leczeniu stosuje się polichemioterapię, radioterapię lub allo-HSCT.
Pierwotny agresywny skórny chłoniak epidermotropowy z cytotoksycznych komórek T CD8+
Pierwotny agresywny skórny chłoniak epidermotropowy z cytotoksycznych komórek T CD8+ przebiega z obecnością zlokalizowanych lub rozsianych grudek, guzków lub blaszek z centralną martwicą i, poza skórą, może zajmować narządy jamy brzusznej, błony śluzowe i układ nerwowy.
Naciek nowotworowy charakteryzuje się obecnością epidermotropowych limfocytów T o fenotypie CD3+, CD8+, bF1+, granzym B+, perforyna+, TIA-1+, CD45RA+, CD45RO–, CD2–, CD4–, CD5–, CD7+/–. W leczeniu stosuje się polichemioterapię, a rokowanie jest niepomyślne.
Pozawęzłowy chłoniak z komórek NK/T typu nosowego
Pozawęzłowy chłoniak z komórek NK/T typu nosowego jest wysoce agresywnym guzem związanym z infekcją EBV. Najczęstszą lokalizacją pozawęzłową tego chłoniaka są jama nosowa i nosogardło. Zmiany skórne obejmują grudki barwy czerwonobrązowej lub wrzodziejące guzki.
W badaniu histopatologicznym stwierdza się naciek komórek NK/T zlokalizowany na różnych poziomach skóry właściwej, zwykle okołonaczyniowo. Rokowanie jest niepomyślne. W leczeniu stosuje się polichemioterapię lub radioterapię.
CHŁONIAKI Z DOJRZAŁYCH KOMÓREK B
Pierwotny skórny chłoniak z ośrodków rozmnażania
Pierwotny skórny chłoniak z ośrodków rozmnażania (follicle center cell lymphoma, FCL) jest nowotworem występującym głównie u pacjentów w wieku podeszłym. Zmiany lokalizują się w obrębie głowy i szyi i mają charakter guzków barwy niebieskoczerwonej otoczonych rumieniowatymi plamami i blaszkami.
W badaniu histopatologicznym skóry stwierdza się proliferację centrocytów i centroblastów w obrębie skóry właściwej. Chłoniak ten cechuje się dobrym rokowaniem (5-letnie przeżycie >95%). W leczeniu pojedynczych ognisk stosuje się leczenie chirurgiczne lub radioterapię, a w przypadku zmian rozlanych lub nawrotu – glikokortykosteroidy miejscowo lub w iniekcjach, IFN-α, rytuksymab lub polichemioterapię.
Pierwotny skórny chłoniak strefy brzeżnej
Pierwotny skórny chłoniak strefy brzeżnej (primary cutaneous marginal zone B-cell lymphoma, PCMZL) jest skórnym odpowiednikiem chłoniak typu MALT i najczęstszym chłoniakiem B-komórkowym skóry. Część przypadków wykazuje związek z infekcją Borrelia burgdorferi lub wirusowym zapaleniem wątroby typu C.
Objawami tego chłoniaka są obecność pojedynczego lub mnogich guzów, które rosną wolno i mogą mieć charakter nawrotowy. W badaniu histopatologicznym stwierdza się obecność nacieków złożonych z limfocytów, komórek plazmatycznych z domieszką centroblastów, komórek limfoplazmocytowych, komórek strefy brzeżnej grudek chłonnych. Rokowanie jest bardzo dobre, lecz w części przypadków choroba przebiega z nawrotami. Leczenie jest podobne jak w przypadku FCL. U chorych z potwierdzonym zakażeniem B. burgdorferi leczenie rozpoczyna się od antybiotykoterapii (penicylina V lub doksycyklina).
Pierwotny skórny chłoniak rozlany z dużych komórek B typu kończynowego
Pierwotny skórny chłoniak rozlany z dużych komórek B (primary cutaneous large B-cell lymphoma, PCLBCL) typu kończynowego dotyka przede wszystkim osoby starsze (7. dekada życia), w większości kobiety. W obrazie klinicznym dominują czerwonobrązowe guzki i blaszki zlokalizowane na kończynach dolnych, ulegające owrzodzeniu.
W badaniu histopatologicznym stwierdza się rozlany naciek złożony z dużych komórek, obejmujący całą szerokość skóry właściwej. Rokowanie w przypadku licznych guzów jest niepomyślne (5-letnie przeżycie 20-55%). Leczenie postaci ograniczonej do skóry obejmuje polichemioterapię z rytuksymabem.
